about Our research
九州大学薬学研究院医薬細胞生化学分野
Department of Cellular Biochemistry
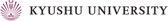
Chromosomal DNA Replication and Cancer Research
In human cells, genomic DNA, which carries genetic information, has to be replicated faithfully, completely, and only once during a single cell cycle to maintain the integrity. If some errors occur during copying DNA (DNA replication), then it would sometimes lead to bad consequences. “Cancer” is one and serious example resulting from such replication errors (mutations).
Molecular mechanisms for cell cycle regulation of DNA replication initiation
Several molecular mechanisms contribute to the maintenance of genomic integrity. Replicative DNA polymerases synthesize new daughter strands using complementary parental strands. However, DNA polymerases rarely incorporate incorrect nucleotides. The mismatch repair pathway that removes inappropriate nucleotides is a fail-safe mechanism for such errors and the disturbance is well known to cause genomic instability and eventual cancer. Chromosomal DNAs are often damaged, for example by ultraviolet, which should also be repaired. Disruption of such repair mechanism also leads to cancer.
We have been interested in elucidating molecular mechanisms for cell cycle regulation of DNA replication initiation, another crucial aspect of replication controls. In human cells, genomic DNA is fragmented into multiple chromosomes, which may allow genome size to expand during the evolution. As a result, DNA replication initiates from multiple replication origins. However, effective operation of the “multiple replication origins” system gives rise to an important problem: i.e. multiple replication origins should each be activated precisely only once during each S phase.
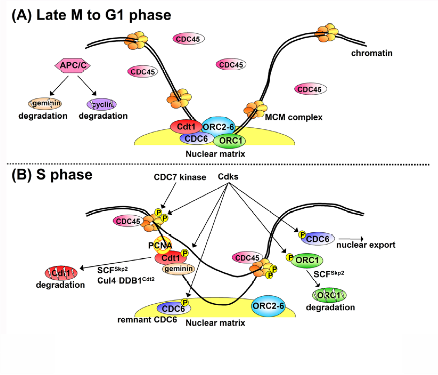
Recent research progress by us and other groups has uncovered the mechanisms (Figure1; Cell Div. 1:22, 2006). It is now clear that the “once and only once replication per single cell cycle” is achieved by the periodic assembly and disassembly of pre-replication complexes (pre-RCs) at replication origins. The pre-RC assembly reaction, known as “licensing”, involves the loading of a replicative helicase, the MCM2-7 complex, onto chromatin by the origin recognition complex (ORC), CDC6 and Cdt1. Two critical inhibitory factors for the pre-RC assembly are cyclin/Cdks (Cdk1 and Cdk2) and geminin. During late mitosis through the G1 phase, a cell cycle regulatory E3 ubiquitin ligase APC/C restrains cyclins and geminin by targeting them for proteolysis through polyubiquitination. Thus, pre-RC assembly only occurs during this period (Figure 1A). Following APC/C inactivation at the onset of S phase, Cdks are activated, stimulating DNA unwinding by MCM (Figure 1B). Then, DNA polymerases synthesize new DNA.
Figure 1. A model for regulation of pre-RC formation during the cell cycle
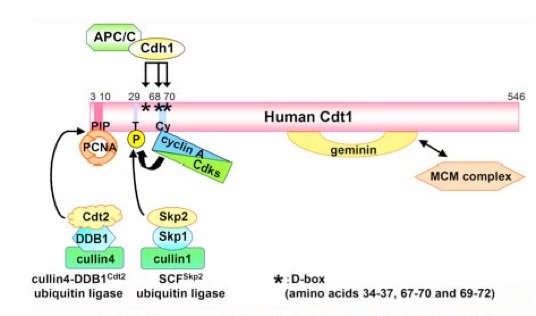
To prevent re-replication, the re-establishment of pre-RC, in other words re-binding of MCM, needs to be suppressed during the S, G2 and M phases of the cell cycle. Cdks play a central role also in this context by preventing re-establishment of pre-RC through multiple mechanisms. One is by phosphorylation of CDC6, leading to CDC6 nuclear export (JBC, 274: 25927, 1999; JBC 277: 10354, 2002; Figure 2). ORC1 is degraded after S phase (JBC, 277: 10354, 2002), presumably depending on phosphorylation by cyclin A/Cdks and binding to SCF-Skp2 ubiquitin ligase.
Figure 2. Nuclear export of human CDC6 protein during the S phase
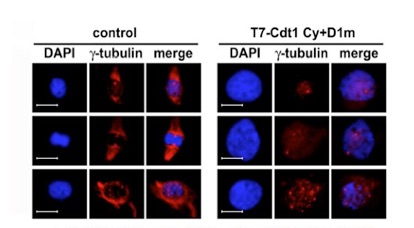
Cdt1, a central factor for the cell cycle regulation of replication initiation: Elucidating the strict regulations by three ubiquitin ligases
It was originally suggested that Cdt1 function is inhibited by geminin binding after S phase. However, we have recently demonstrated that three ubiquitin ligases strictly control Cdt1 proteolysis (Figure 3), showing that Cdt1 is a central player in the cell cycle regulation of replication initiation. During S and G2 phases, Cdt1 is brought to proteolysis by Cdk phosphorylation-dependent SCF-Skp2-mediated ubiquitination (JBC 279: 19691, 2004). Interestingly, Cdt1 is also regulated by replication-coupled, Cullin4-DDB1-Cdt2 ubiquitin ligase-mediated ubiquitination, which is dependent on Cdt1 binding to PCNA, an eukaryotic sliding clamp stimulating DNA polymerases (EMBO J 25: 1126, 2006). In addition, when cells enter quiescence, Cdt1 is rapidly cleared by APC/C-Cdh1-mediated proteolysis (MBC 19: 1007, 2008).
Figure 3. Molecular mechanisms for Cdt1 proteolytic regulations by three ubiquitin ligases
Cdt1 deregulation induces chromosomal instability, a mechanism leading to cancer
As expected from the above strict regulations, deregulation of Cdt1 is a deleterious insult, leading to re-replication and/or chromosomal damage (JCS 119: 3128, 2006; MBC 19: 1007, 2008; JCS 122: 1184, 2009; Figure 4). The induced chromosomal instability may eventually lead to carcinogenesis and Cdt1 overexpression is in fact often observed in human cancers (JCS 119: 3128, 2006). By other groups, it has been suggested that Cdt1 overexpression could endow cells with the transforming ability.
Figure 4. Nuclear enlargement upon rereplication induced by overexpression of a proteolysis-resistant mutant Cdt1
Cdt1-geminin system could be a novel molecular target for anti-cancer chemotherapeutic agents
Cdt1 overexpression can induce remarkable rereplication in cancer-derived cells but not in non-transformed cells (JCS 122: 1184, 2009). We therefore think that tumor cells could be selectively eliminated by modulating the Cdt1-geminin interactions and have been seeking small molecule compounds that affect the interaction. During the screening of many compounds, we have found some interesting ones that can selectively kill cancer-derived cells in vitro and/or can inhibit Cdt1-geminin binding in vitro. Using these lead compounds, we are now trying to develop novel anti-cancer drug.
Future research focuses: Involvement of replication licensing factors in other chromatin regulations.
For details, please see our recent papers (Genes Cells 13, 1045, 2008; JCS 123: 225, 2010; JBC 286: 39200, 2011).

